The struggle between viruses and humans has been a protracted war spanning the course of life’s evolution. Viruses constantly assault the human body, causing numerous diseases, while the immune system fights back and gradually incorporates viral sequences into the genome, resulting in co-evolution. Endogenous retroviruses (ERVs) are “fossils” left in the chromosomes by ancient retroviruses that invaded human cells millions of years ago. They now make up about 8% of the human genome, leaving a unique genetic imprint. Most ERVs remain silent under epigenetic and genomic erasure mechanisms, serving as “dark matter” within the genome. Elucidating their physiological and pathological significance has long been a grand challenge in life sciences.
Heart failure (HF) is a complex clinical syndrome characterized by the inability of the heart to pump sufficient blood to meet the body’s needs. Globally, an estimated 164 million people suffer from HF. Due to the lack of effective treatments, its 5-year mortality rate exceeds 50%, higher than that of most cancers, making it a leading cause of death worldwide.

On August 18, the team of Professors ZHANG Bing, SUN Kun, and ZHU Dan from SJTUSM published a study in Circulation titled An Aberrant Resurgence of Endogenous Retroviruses Prompts Myocarditis and Heart Failure. This work was the first to demonstrate that reactivation of endogenous retroviruses, particularly the ERV1 subfamily, occurs across multiple types of HF. Reactivated ERVs trigger innate immune signaling through Toll-like receptors TLR7/9, thereby promoting myocarditis and HF. Conversely, suppressing ERV reactivation or blocking excessive innate immune activation significantly alleviates HF.
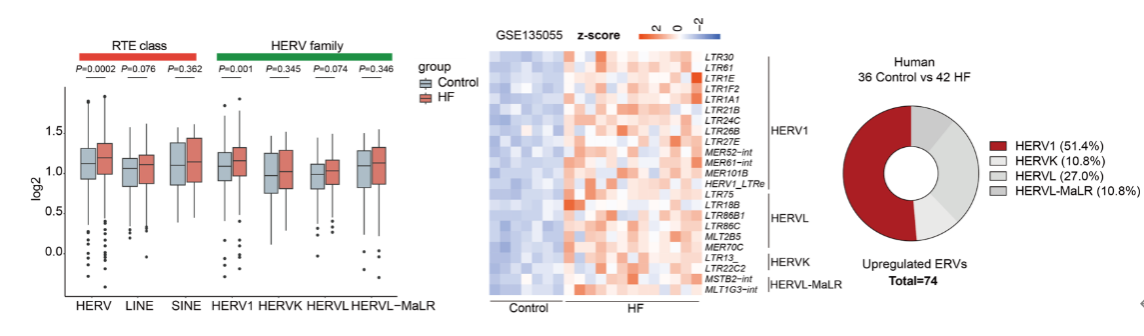
To explore the role of ERVs in HF, the authors first developed an analytical method for retrotransposon RNA and systematically analyzed ventricular RNA-seq datasets from 78 published patients with dilated cardiomyopathy (DCM) and ischemic cardiomyopathy (ICM). They found that ERVs were significantly upregulated in failing myocardium, whereas other retrotransposons such as LINEs and SINEs showed no notable changes. Among ERV subfamilies, HERV1 was consistently and markedly upregulated across all datasets, emerging as the predominant activated subtype. The activation of HERV1 was further confirmed in both mouse and non-human primate HF models, indicating that ERV1 activation represents a common molecular signature of HF.
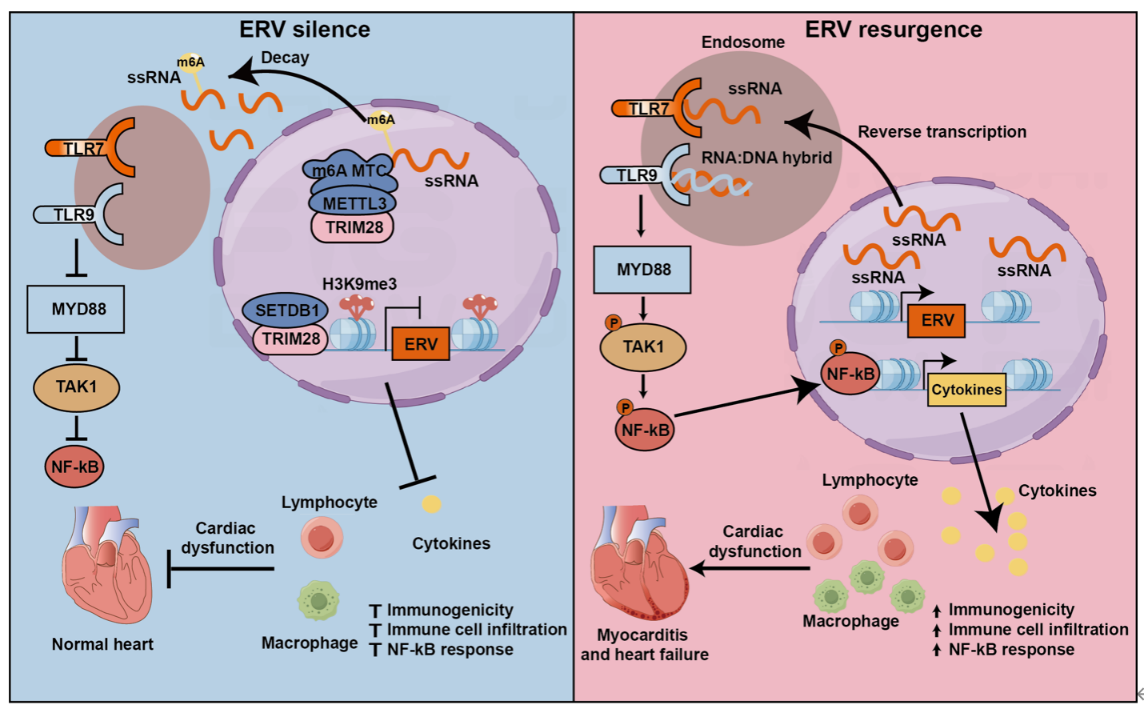
Epigenetic repression is the major mechanism silencing genomic ERVs. This study revealed that TRIM28, a key epigenetic suppressor, was markedly downregulated in HF myocardial samples. To investigate its regulatory role, the team generated TRIM28 conditional knockout mice. Transcriptomic analysis showed a pronounced upregulation of ERV genes, dominated by ERV1. Fluorescence in situ hybridization confirmed cytoplasmic punctate accumulation of ERV1 in cardiomyocytes, consistent with activation patterns observed in HF models. TRIM28 deficiency rapidly led to severe acute HF phenotypes—marked decline in cardiac function, chamber dilation, wall thinning, elevated HF biomarkers, and pulmonary/hepatic congestion and edema. Mice began dying as early as day 4 post-knockout.
Mechanistic studies revealed that TRIM28 restrains ERV activation through dual epigenetic mechanisms. On one hand, TRIM28 enforces H3K9me3 histone modification to silence certain ERVs; on the other, for ERVs not regulated by H3K9me3, TRIM28 recruits the m6A methyltransferase METTL3 to modify and degrade ERV RNAs, thereby suppressing their expression. Activated ERVs in cardiomyocytes triggered a robust innate immune response, with their RNA transcripts or reverse-transcribed DNA recognized by TLR7/9, leading to specific activation of the NF-κB inflammatory pathway and downstream cytokines IL-6, IL-1β, and TNF-α. Interestingly, unlike exogenous viruses that typically stimulate interferon (IFN) pathways, ERV activation did not trigger the IRF–IFN axis but specifically activated NF-κB. Aberrant innate immune activation drove infiltration of CD3+ T cells and CD68+ macrophages, causing myocardial inflammation, cardiomyocyte death, and ultimately acute HF. Time-course analyses showed that ERV activation and inflammation preceded the rise of HF biomarkers, confirming ERV activation as an initiating event in myocarditis and HF.
Therapeutic intervention studies demonstrated that inhibition of TLR7/9 with NSC4375 or NF-κB with JSH23 markedly improved survival of TRIM28-deficient mice, reduced inflammatory infiltration and fibrosis, and preserved cardiac function. Reverse transcriptase inhibitors produced similar cardioprotective effects, further confirming the pathogenic role of ERVs. Acute HF (AHF)—the most severe subtype of HF, including sudden-onset HF or rapid decompensation of chronic HF—has a one-year mortality of ~30%. Myocarditis, particularly fulminant myocarditis, is a common underlying cause of AHF and is usually triggered by exogenous viruses such as coxsackievirus B, adenovirus, herpesvirus, HIV, and SARS-CoV-2. This study is the first to demonstrate that ERV reactivation can also cause acute myocarditis and HF. Ischemia–reperfusion (I/R) injury is a common cause of HF. In an I/R HF model, the authors observed significant activation of the TRIM28–ERV–TLR7/9–NF-κB axis, suggesting that targeting this pathway may confer protection. Restoring TRIM28 specifically in cardiomyocytes via AAV9 delivery effectively suppressed ERV activation, blocked TLR7/9–NF-κB signaling, and reduced pro-inflammatory cytokine expression. AAV9-TRIM28 treatment significantly reduced infarct size and improved cardiac function and remodeling. Similarly, inhibition of innate immunity with TLR7/9 inhibitor NSC4375 achieved protective effects. Notably, collaborators at Huazhong University of Science and Technology (Professors WANG Daowen and CHEN Chen) and other teams previously reported that TLR7/9 inhibitors alleviated myocarditis and HF caused by SARS-CoV-2 infection during the COVID-19 pandemic. Together, these findings underscore the central pathogenic role of TLR7/9-mediated innate immune activation in myocarditis and HF.
In summary, this study is the first to demonstrate that ERV activation, particularly the ERV1 subfamily, is a common molecular and pathological feature of HF. It also identified a novel signaling axis, the TRIM28–ERV–TLR7/9–NF-κB pathway, and established its critical role in myocarditis and HF. Targeting this pathway may provide new therapeutic opportunities for treating myocarditis and HF.
This work was supported by the Ministry of Science and Technology, the National Natural Science Foundation of China, Shanghai Science and Technology Commission, Shanghai Municipal Education Commission, and Shanghai Jiao Tong University “Star Program.” Professors ZHANG Bing and SUN Kun of Xinhua Hospital, and Professor ZHU Dan of Shanghai Chest Hospital, all affiliated with Shanghai Jiao Tong University School of Medicine, served as corresponding authors. XIONG Junhao (PhD candidate, Institute of Systems Biomedicine, SJTU), ZHANG Shasha (Assistant Researcher, SJTU), GENG Zilong (PhD candidate, SJTU), and LIN Juntao (Master’s student, SJTU) were co–first authors. The study also received collaborative support from Professors SUN Aijun (Zhongshan Hospital, Fudan University), WU Gengze and ZENG Chunyu (Third Military Medical University), CHEN Chen and WANG Daowen (Huazhong University of Science and Technology), ZHANG Yan (SJTU School of Biomedical Engineering), CHEN Fengyuan, HOU Xumin, and LI Ruogu (SJTU School of Medicine), GUO Xiaoling (Wenzhou Medical University), FENG Yuliang (Southern University of Science and Technology), TIAN Xiaoyu (Chinese University of Hong Kong), CHEN Jiekai (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences), and YI Chengqi (Peking University).
