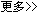创新药物研发中活性分子(苗头分子hit/先导分子lead)的发现是新药研发的源头。传统活性化合物的发现主要依赖高通量筛选,但受限于现有化合物库的库存(仅数亿个,10^9次方)和多样性,发现具有全局优势的药物分子几率低下。相较之下,从全局化学空间中从头(de novo)分子设计,即在10^50次方的全局化学空间中“从无到有”创造活性分子,极大地提升了全局优势药物分子发现的可能性。

近日,来自上海交通大学医学人工智能研究院/基础医学院药物学与人工智能交叉学系的张健课题组在Nature Machine Intelligence在线发表题为“Electron-density-informed effective and reliable de novo molecular design and optimization with ED2Mol”的研究论文,报道了自主研发的人工智能药物设计技术——ED2Mol (transformingElectronDensityTobioactiveMolecules )。该工作建立了从苗条分子生成到先导分子优化的统一智能框架,自动化生成的活性分子在分子间和分子内可靠性上实现领域最优,面向4个重大疾病靶标设计并优化出多类的活性分子,不仅在常规正构位点上表现出色,在一直难以突破的变构位点分子生成上也获得突破,发现了多类抑制剂与激动剂等,为创新药物发现提供了高效且可靠的设计平台。由于该工作对人工智能药物设计领域的重要意义,被该期刊选为封面故事推荐。

从原理上看,结构生物学通常借助配体电子密度图进行精确结构解析:配体电子密度在宏观上表征配体—受体的形状互补,在微观上编码统一的电磁相互作用。将其显式融入深度生成模型,既可把握结合口袋的全局互补环境,又能更通用且精确地建模原子级相互作用。在实现路径上,ED2Mol首先利用变分自编码器根据结合口袋结构预测配体的电子密度;随后采用分层峰度搜索算法拟合核心骨架;最后通过等变图神经网络将分子片段迭代式延展至尚未被占据的电子密度区域。基于同一思想,ED2Mol亦可便捷用于苗头化合物优化:在电子密度的约束与引导下,从预定义的苗头核心出发,分段扩展并生成效力更强的候选分子。
在多个基准上的广泛评估表明,ED2Mol生成的化合物在结合效能、结合位姿的质量与稳定性方面全面领先,同时具备优良的可合成性、类药性和结构多样性。面对更具挑战性的、此前未见的变构口袋基准,ED2Mol仍展现出良好的泛化能力。为验证其应用潜力,研究团队在5个真实世界任务中开展设计与优化:FGFR3正构抑制剂的从头生成与优化、CDC42变构抑制剂的从头生成、GCK变构激活剂的优化,以及GPRC5A变构调节剂的从头生成。上述任务覆盖正构与变构位点,并涉及抑制剂与激动剂等多类配体,体现出该方法在多样化药物发现场景中的通用性。此外,作者还通过X射线衍射等结构生物学手段验证了生成分子与靶蛋白的结合模式。
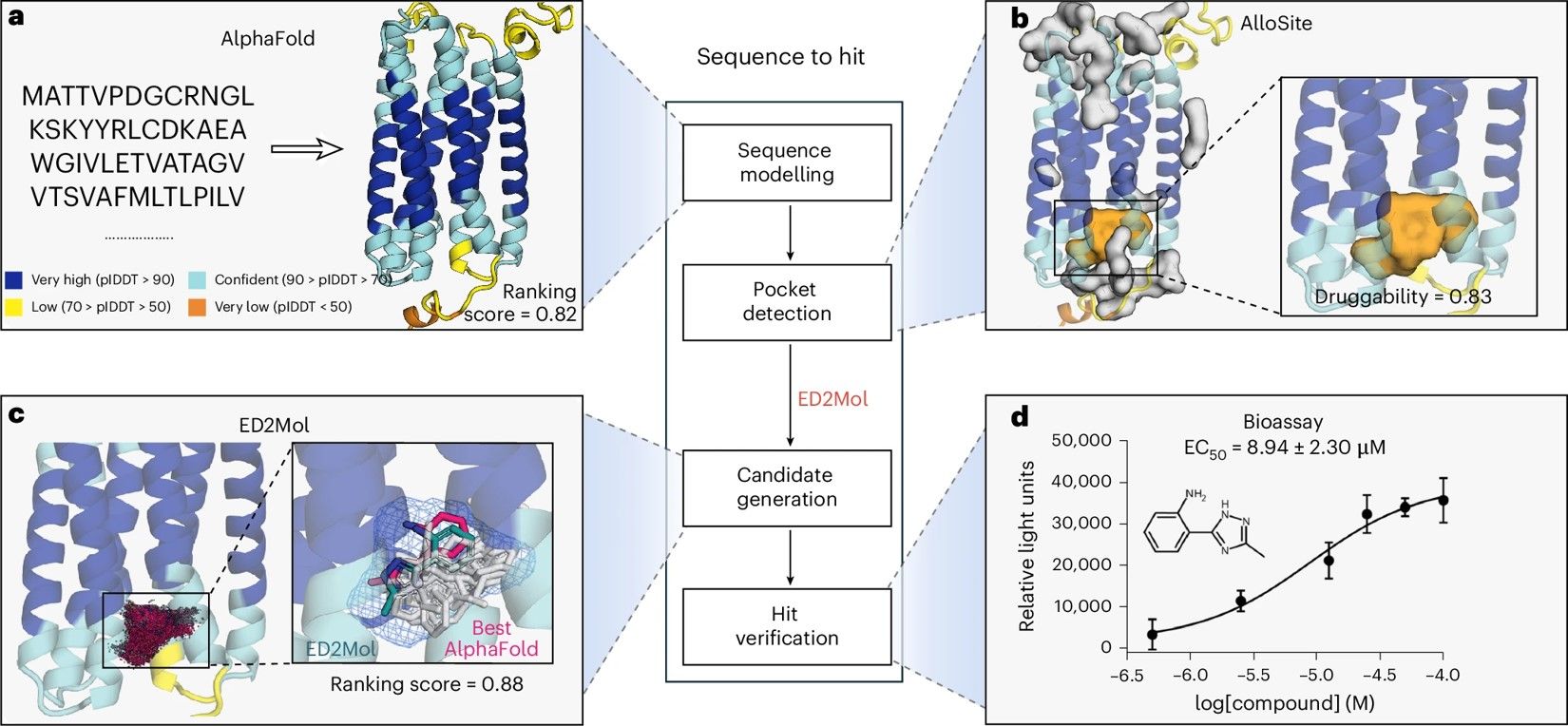
特别值得一提的是,ED2Mol在孤儿受体GPRC5A上实现了变构调节剂的从头设计。研究团队构建了“序列到苗头(sequence-to-hit)”框架,整合序列到结构建模(AlphaFold)、结构到口袋挖掘(AlloSite)、口袋到候选生成(ED2Mol)以及候选到苗头验证(PRESTO-Tango)等环节,最终发现全新GPRC5A激动剂A4,为开发高效GPRC5A激活剂迈出了关键一步。
本项研究建立了人工智能产学研合作的新范式,由上海交通大学医学人工智能研究院/基础医学院药物学与人工智能交叉学系的张健教授领衔(通讯作者),上海交通大学博士研究生李明玉、宇道生物宋堃博士、上海交通大学博士研究生何继骁为共同第一作者,宇道生物、青煜医药2家创新biotech共同研发、并获中国科学院上海药物研究所研究员赵玉军的大力支持。相关工作获BT-IT国家重点研发计划、国家自然科学基金重点项目、上海重点创新团队(药学)、繁星基金会、临港实验室、博士生科创培育基金等项目支持。