疼痛管理是全球医疗卫生领域面临的重要挑战。对于中重度慢性疼痛患者而言,长期疼痛不仅严重影响生活质量,也给社会和医疗系统带来沉重负担。阿片类药物仍是临床镇痛的核心手段,然而现有主流镇痛药物大多靶向μ型阿片受体(μOR),在发挥强效镇痛作用的同时,伴随成瘾、呼吸抑制等严重不良反应,由此引发的阿片药物滥用危机已成为全球公共卫生问题。因此,开发兼具高效镇痛作用和良好安全性的替代靶点与新型药物,是当前镇痛药物研发的重要方向。κ型阿片受体(κOR)因其独特的镇痛机制与相对较低的成瘾风险,被视为极具潜力的新一代镇痛靶点。近年来研究通过生化与单分子成像手段发现,相较于其他阿片受体亚型,κOR在生理表达水平下即可于低受体密度条件下自发形成稳定的同源二聚体,具有显著更高的二聚化倾向1, 2。然而,κOR二聚体究竟如何组装形成、膜脂质在其中发挥何种作用,以及二聚化如何影响受体信号转导,长期以来缺乏明确的结构与机制解释,限制了基于κOR二聚体的新药设计。
2026年5月29日,上海交通大学庄友文团队联合华中科技大学刘剑峰团队、中国科学院上海药物研究所徐华强团队,在Nature Communications在线发表了题为“Structural characterization of kappa-opioid receptor dimer in complex with two G proteins”的研究成果。该研究综合运用活细胞成像、冷冻电镜以及多种生化与药理学技术手段,在活细胞层面直接证实了κOR稳定同源二聚体的存在,解析了天然产物激动剂Salvinorin A(SalA)结合的κOR单体及二聚体分别与Gi蛋白形成复合物的高分辨率三维结构,深入阐明了膜脂质介导的二聚体组装机制、SalA非经典激活κOR及亚型选择性识别机制,以及受体二聚化通过双Gi蛋白同步偶联放大下游信号的全新模式,为开发安全高效的κOR靶向镇痛新药提供了关键分子结构依据与药理学理论支撑。
·活细胞证实κOR稳定二聚化
在本项研究中,研究团队首先利用双分子荧光互补(BiFC)实验,在活细胞中直接验证了κOR同源二聚体的存在。分别携带Venus荧光蛋白VN与VC片段的两种κOR构建体共表达时,可产生明显的膜定位荧光信号;单独表达任一构建体或引入无关膜蛋白均不产生或不影响信号,证明κOR二聚化具有高度特异性。进一步通过NanoBiT互补发光实验验证,多种κOR激动剂处理均未能完全破坏二聚体的形成,表明κOR二聚化是一种配体非依赖性的稳定相互作用。
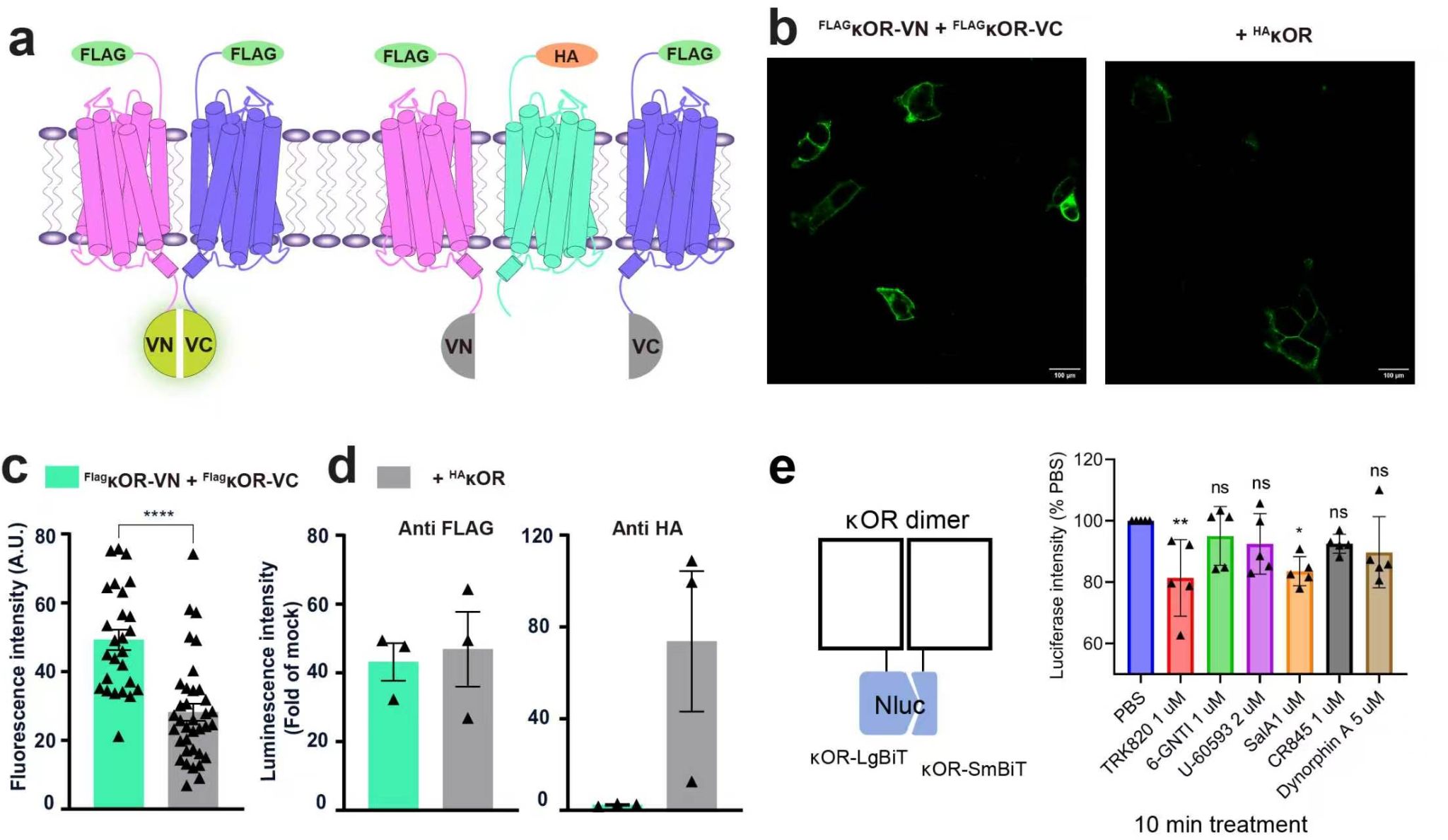
图1. κOR二聚化的BiFC图像和介导的下游信号通路。a: BiFC实验的示意图, b: HEK293细胞的代表性BiFC图像, c: 膜BiFC荧光强度的定量分析, d: FLAG标记和HA标记κOR的膜表达水平, e: BRET实验检测G蛋白募集到κOR二聚体的示意图及SalA诱导的G蛋白募集检测结果。
·冷冻电镜解析KOR-Gi信号复合物单体与二聚体的三维结构
随后,研究团队在体外表达组装了不同聚集形态的KOR-Gi信号复合物,并利用冷冻电镜技术分别解析了SalA激活下κOR-Gi单体与二聚体信号复合物的结构,整体分辨率分别为3.2 Å和3.6 Å。二聚体结构揭示,两个κOR原体呈平行镜像排列,每个原体各自偶联一个Gi蛋白,形成2:2的化学计量比构型。这一“双G蛋白耦联”模式有别于此前报道的GPR3和APJ等多种GPCR二聚体3, 4,后者通常每个二聚体单元仅偶联一个G蛋白,代表了A类GPCR二聚体信号转导的一种全新构型。功能实验进一步证实,任一原体的G蛋白偶联能力受损均会显著降低整体信号强度,说明κOR二聚体并非简单的聚集,而是通过双Gi蛋白同步耦联实现协同信号放大,从而显著增强下游信号转导效率。
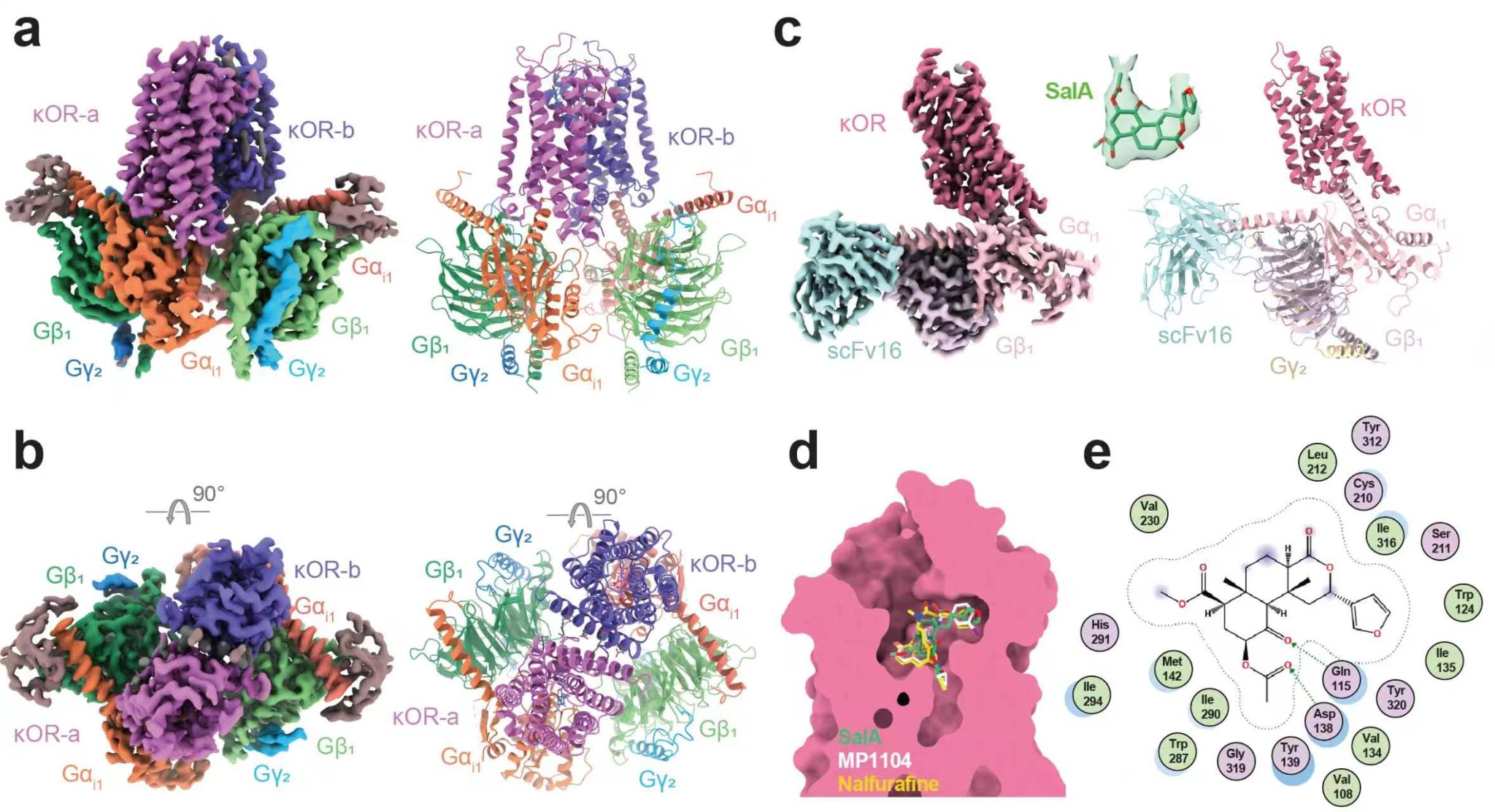
图2. SalA结合κOR–Gi二聚体及单体复合物的冷冻电镜结构。 a–b: SalA–κOR–Gi的二聚体结构, c: SalA–κOR–Gi的单体结构, d: SalA, MP1104和nalfurafine在κOR正构口袋的结合模式, e: SalA与κOR结合口袋氨基酸相互作用的二维示意图。
·SalA非经典激活机制与受体亚型选择性
值得注意的是,SalA是一种来源于鼠尾草属植物的天然二萜类化合物,与传统阿片类配体不同,其结构中不含经典阿片药物普遍具有的与受体D1383.32形成盐桥的碱性氮原子。研究显示,SalA的萜环羰基氧与乙酰氧基通过氢键与κOR正性结合口袋中的D1383.32、Q1152.60和Y3207.43等残基组成极性相互作用网络,并结合广泛的疏水作用,在缺乏传统盐桥锚定的情况下仍能实现对κOR的高亲和力结合和高效激活,从而揭示了一种区别于经典阿片药物的受体激活模式,为设计新型非经典阿片配体提供了重要模板。研究通过突变实验进一步鉴定出Y3127.35是决定SalA对κOR亚型选择性的关键残基:在μOR、δOR和NOPR的对应位置分别为色氨酸或亮氨酸,无法与SalA的二萜基团形成有效相互作用;将上述受体该位点替换为酪氨酸后,SalA效价均提升约5-10倍,反之将κOR该位点突变则显著降低SalA效价,而不影响内源性配体强啡肽的活性,表明Y3127.35是SalA选择性识别κOR的特异性决定因子。
·脂质介导的二聚体界面
研究显示,κOR的二聚界面主要由跨膜螺旋TM1与胞内螺旋8(Helix 8)介导,界面处分布有多个脂肪酸和胆固醇分子,但界面接触面积(469 Å2)显著小于已报道的多种TM1-TM1界面GPCR二聚体。研究团队通过NanoBiT滴定实验与丙氨酸突变扫描发现,V751.48、F3418.54和F3468.59等关键疏水残基直接参与原体间接触,对二聚体稳定性贡献最为显著;而界面脂质则充当动态“分子填充物”,桥接两个原体的疏水表面,弥补相对有限的蛋白质直接接触面积。此外,通过甲基-β-环糊精(MβCD)急性耗竭细胞膜胆固醇,κOR二聚化信号显著降低;而在胆固醇接触残基突变体中,上述耗竭效应消失,直接证明界面胆固醇对κOR二聚体稳定具有特异性功能贡献,而非仅是膜环境的非特异性影响。
该研究首次从结构与功能两个层面系统阐明了κOR同源二聚体的组装机制及其信号放大功能,揭示了A类GPCR同源二聚体可同时偶联两组G蛋白的新型信号转导模式,并为理解膜脂质参与GPCR二聚体调控提供了直接证据。上述发现不仅深化了阿片受体信号转导的基础认知,也为靶向κOR二聚体界面或利用其信号增强特性开发下一代非成瘾性镇痛药物提供了重要的理论依据。
本研究冷冻电镜数据由上海市高峰电镜中心完成收集;上海药物所/南京中医药大学联合培养博士生赵雨茜、华中科技大学许婵娟副研究员、上海药物所王悦副研究员为论文的共同第一作者。上海交通大学庄友文研究员、华中科技大学刘剑峰研究员、上海药物所徐华强研究员为论文的共同通讯作者。参与本次研究的还有北卡罗来纳大学教堂山分校Bryan L. Roth院士、Xi-Ping Huang博士和Jing Wang博士等。该工作获得了国家自然科学基金委、科技部重点研发计划、上海市科技重大专项/面上以及何享健青年科学家项目等经费资助。
原文链接: https://www.nature.com/articles/s41467-026-73615-x
参考文献:
1. Cechova, K. et al. Kappa but not delta or mu opioid receptors form homodimers at low membrane densities. Cell Mol Life Sci 78, 7557-7568 (2021).
2. Zhou, P. et al. Single-molecule detection of transient dimerization of opioid receptors 1: Homodimers’ effect on signaling and internalization. BioRxiv (2024).
3. Yue, Y. et al. Structural insight into apelin receptor-G protein stoichiometry. Nature structural & molecular biology 29, 688-697 (2022).
4. Chang, H. et al. Structural basis of oligomerization-modulated activation and autoinhibition of orphan receptor GPR3. Cell reports 44, 115478 (2025).