12月9日, 基础医学院王锋课题组在Immunity杂志在线发表了题为“The TCR-SUB1-DOCK2 Axis Promotes Autoimmunity by Driving Pathogenic CD4⁺ T Cell Tissue Infiltration”的研究论文。该研究系统阐明了TCR信号途径下游关键分子SUB1通过定量调控Dock2基因转录, 驱动炎性CD4⁺ T细胞向病变组织浸润,从而诱发自身免疫疾病的全新机制。研究揭示SUB1作为调控致病性CD4⁺ T细胞组织迁移的“分子守门人”, 为精准靶向免疫细胞迁移,治疗自身免疫疾病提供了潜在新策略。

自身免疫疾病发生发展的核心环节之一是活化炎性T细胞向特定组织的异常迁移与浸润, 因此阻断该过程已成为极具前景的治疗策略之一1-4。细胞迁移依赖细胞骨架动态重组, 为细胞运动提供所需的机械力5, 6。然而, 从T细胞受体 (TCR) 识别抗原到定量驱动细胞骨架重排、最终决定T细胞组织浸润能力的关键调节因子及信号通路, 迄今仍未阐明。
基于前期临床数据的分析, 本研究团队发现SUB1在多种自身免疫病患者CD4⁺ T细胞中表达显著上调, 且其表达受TCR-IRF4信号轴直接调控。研究人员通过构建T细胞SUB1特异性敲除小鼠并诱导实验性自身免疫性脑脊髓炎 (EAE) ,发现SUB1缺失可完全防止EAE发病, 并显著减少CD4⁺ T细胞的中枢神经系统 (CNS) 浸润。进一步整合多组学分析表明, SUB1缺失导致关键迁移调节因子DOCK2表达下调,进而抑制Rac依赖的肌动蛋白聚合, 最终削弱T细胞的迁移能力。深入的分子机制研究揭示, SUB1可通过液-液相分离 (LLPS) 形成生物分子凝聚体以打开特定染色质区域, 进而直接反式激活Junb转录, 并与JUNB蛋白结合协同放大Dock2基因表达 (图1左)。
值得注意的是, CD4⁺ T细胞的SUB1缺失仅使DOCK2表达量下降约50%, 因此SUB1轴对DOCK2的定量调控特性为自身免疫疾病的精准干预提供了重要契机: 靶向SUB1可能实现一个最佳治疗窗口, 在特异性阻断致病性CD4⁺ T细胞向炎症组织 (包括中枢神经系统、肠道及皮肤等) 浸润的同时, 最大程度地保留T细胞淋巴器官迁移生理功能及免疫防御作用 (图1右)。综上, 本研究阐明了一条连接TCR抗原识别信号与T细胞组织迁移的关键分子通路—TCR-SUB1-DOCK2轴, 不仅深化了对自身免疫疾病发病机制的理解,也为开发聚焦细胞迁移途径的精准、安全免疫干预策略,提供了新的理论依据与潜在靶点分子。
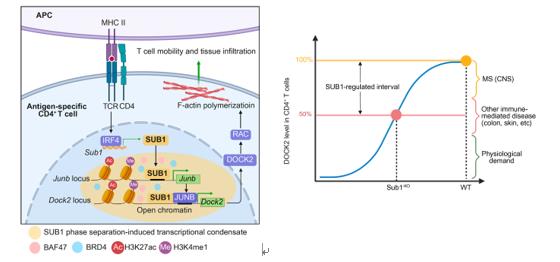
图1. TCR-SUB1-DOCK2通路调控CD4⁺ T细胞
组织浸润及自身免疫疾病机制
基础医学院博士生李晓雪, 博士后梁文华和科研助理王为芳为文章的共同第一作者, 王锋研究员为文章的独立通讯作者。该研究工作得到国家自然科学基金重点项目, 国家重点研发计划, 上海市科学技术委员会, 上海市地方高水平大学协同创新团队和上海市免疫学研究所卓越科学家项目等经费支持。