
The focus of Dr. Jan Vijg’s research interests is the causal relationship between somatic DNA mutations, aging and age-related diseases. His research has led to the development and application of mouse and Drosophila models for studying somatic mutations in vivo. More recently, his lab in New York has developed single-cell whole genome sequencing methods for quantitatively analyzing genome-wide mutations in primary human cells. At the CSCOmics his research is focused on the application of these methods to study the possible causal relationship between somatic mutations and two common diseases: atherosclerosis and osteoarthritis.
Somatic mutations in age-related diseases other than cancer
DNA mutations in somatic cells are key causal factors in cancer initiation and progression. Age-related accumulation of somatic mutations in normal cells have been implicated in the increased cancer risk with age (DePinho, 2000; Vijg, 2014), which steeply rises after age 40. Advancing age is the most important risk factor for cancer overall, and for many individual cancer types (https://seer.cancer.gov/). However, there is also evidence that somatic mutations could underlie other age-related, chronic diseases, such as cardiovascular disease, arthritis, diabetes and neurodegenerative diseases (Erickson, 2003; Poduri et al., 2013). This area of research is driven by the enormous increase of genomic sequencing in studying single gene disorders and there are now many examples of somatic mosaicism as a cause of diseases hitherto only known as caused by germ line mutations. This is mostly limited to diseases where such mosaicism can be easily seen, for example, neurofibromatosis I (Erickson, 2003). However, there is at least one case in which Alzheimer’s Disease is apparently caused by somatic mosaicism for a mutation in the presenilin-1 gene(Beck et al., 2004).
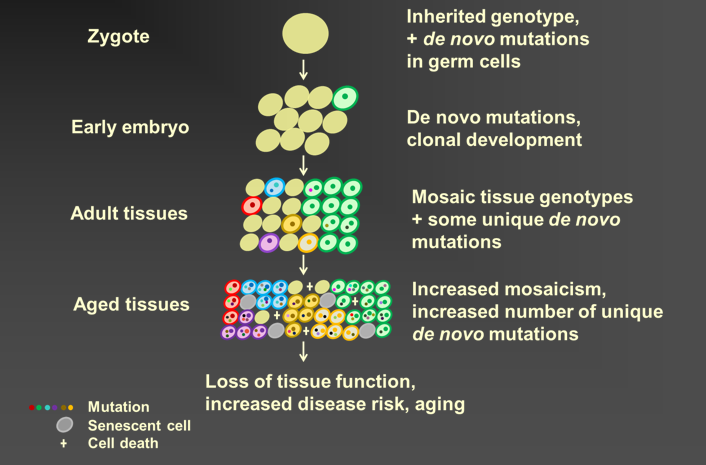
Somatic genome mosaicism
There are at least two mechanisms through which somatic mutations can cause disease. The first is by a combination of cell growth and selection, similar to how mutations cause cancer. This is the mechanism that will be studied by my lab at CSCOmics and it applies best to diseases that have been associated will cell growth, caused mostly by increased inflammation. The second mechanism is simply increased mutation loads of cell in vivo. This mechanism applies best to aging in general and is currently studied extensively by my lab at Einstein. The single-cell, mutation analysis methodology, developed for that purpose, will be used at CSCOmics as well.
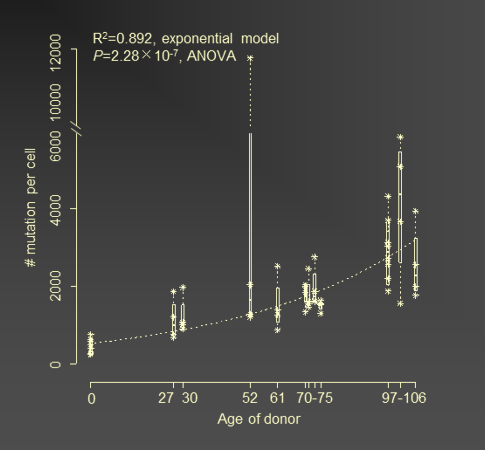
Mutations in human B lymphocytes accumulate exponentially with age
The genetic causes of complex, multifactorial heritable diseases are difficult to unravel because of the large number of DNA sequence variants, often rare, variants, that can be involved in such diseases. Recent progress in massive whole genome and whole exome sequencing has provided a wealth of potentially causal genetic variants and the problem now is to functionalize these variants to unravel the disease mechanism and discover genetic targets that can provide treatment. The situation of a somatic mutational cause for such diseases is even more of a challenge. Indeed, sequencing of bulk DNA isolated from human tissues will not reveal the rare, low-abundant mutations that could be the cause of such diseases. It is for this reason that a single cell approach is necessary. In my lab at Einstein we developed a highly accurate single-cell whole genome sequencing approach that allows the identification of most potentially causal genetic variants, from base substitutions and insertions/deletions (indels), to copy number variants, retrotranspositions and genome structural variations (Dong et al., 2017). At least one other group developed such an assay, but only for analyzing a much smaller part of the genome (Lodato et al., 2018). Using this method a surprisingly high number of base substitution mutations was reported for different types of human neurons. Also other types of mutations have been found in single human primary cells, but a systematic study is thus far lacking (Zhang and Vijg, 2018). At CSCOmics I plan to study two chronic, age-related disorders: cardiovascular disease and osteoarthritis.
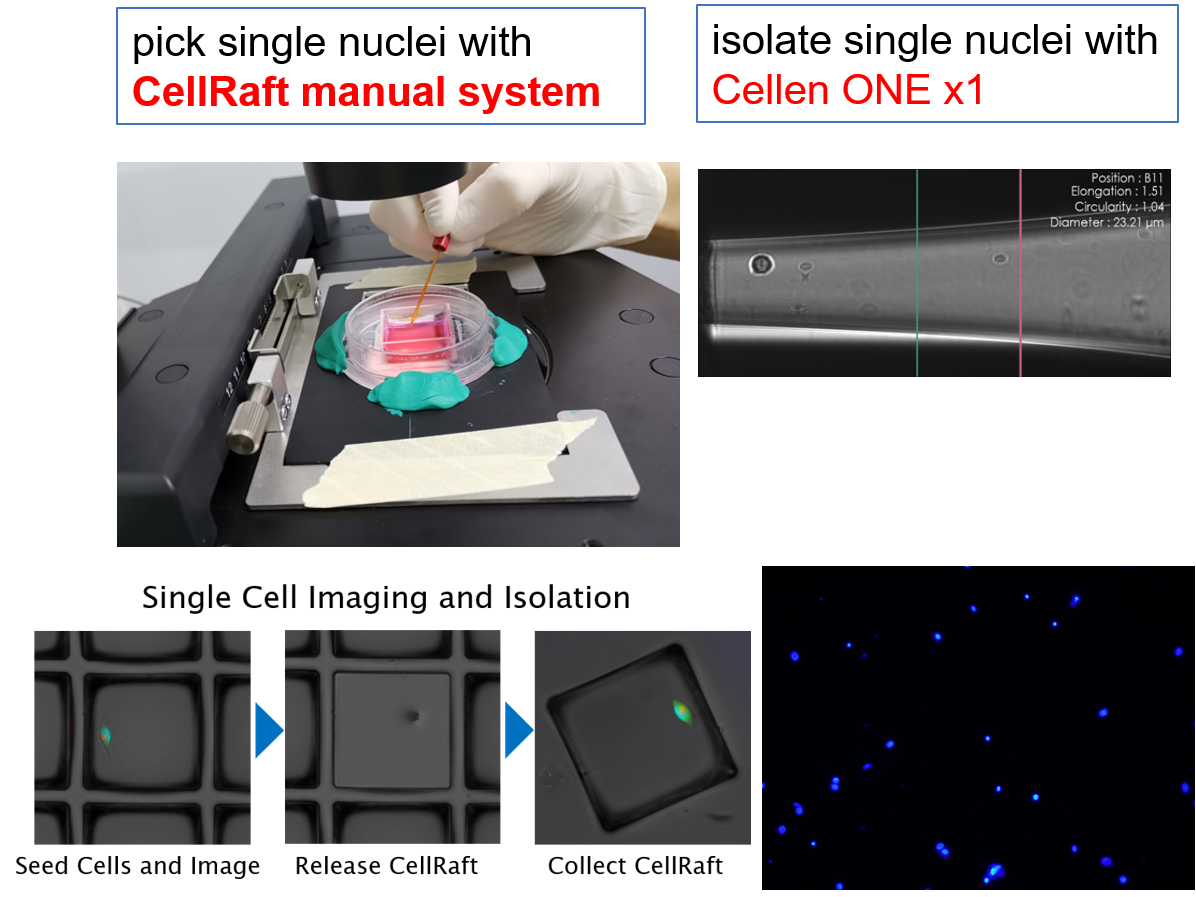
Single nuclei from frozen tissue sample
Cardiovascular Disease (CD)
CD remains one of the two main causes of death in both the US and China (WHO) in spite of recent progress in preventative measures. The main cause of cardiovascular disease is atherosclerosis, characterized by a buildup of plaque in arteries, which constrains blood flow. There is now ample evidence that atherosclerotic lesions show clonal cell growth and, therefore have been hypothesized to be neoplastic processes, arising through critical somatic mutations. This process of aberrant clonal cell growth in blood vessels is likely driven by chronic inflammation (Fuster et al., 2017; Weakley et al., 2010). Due to the lack of the necessary single-cell mutation analysis technology, there is very limited evidence for actual de novo mutations in atherosclerotic lesions. Exceptions are mutations in the relatively unstable microsatellite loci. Somatic mutations have been detected in the A10 microsatellite of the TGFBR2 gene in atherosclerotic plaques of human coronary arteries (McCaffrey et al., 1997).
I will obtain atherosclerotic plaques from carotid arteries of patients undergoing endarterectomy and isolate cells as described (Lebedeva et al., 2017). Part of the plaque material will be used for histochemistry. Plaque cells will be analyzed, first by flow cytometry (Bonanno et al., 2000) and subsequently sorted into small PCR tubes for whole genome amplification and sequencing as described (Dong et al., 2017). Sequencing data will be analyzed for various types of mutations.
Osteoarthritis (OA)
Osteoarthritis (OA) is a devastating, but very common age-related disease, with an estimated prevalence of more than 60 million people in the United States and well over 100 million in China in people aged 60 years and over. The etiology and the pathogenetic processes for the majority of OA cases remains unclear. Like for atherosclerosis also for OA somatic mutagenesis has been implicated as a possible cause of the disease. Cytogenetic studies have shown that in cells from affected knee samples increased chromosomal aberrations appear early and may be a causal factor in the disease (Mertens et al., 1996). Likewise, we will isolate cells from affected knee samples and subject them to our single-cell whole genome analysis method to study the landscape of all types of somatic genome alterations quantitatively.
Other age-related diseases
Other age-related disease that could be due, at least in part, to somatic mutagenesis are fibrosis and neurodegenerative diseases (e.g., Alzheimer’s disease and Parkinson’s disease). We could use the same strategy as described above to study lesions or their surrounding regions for somatic mutations. This certainly is a future plan but depends to some extent of what will be found with CD and OA.
Mechanisms
While the work described above will provide the first quantitative information as to whether somatic mutagenesis is in fact a possible component of the etiology of age-related disease, it does not address the mechanism. Once we will have established that mutation load in cells of these pathological lesions is indeed elevated, we will analyze the nature of the mutations (mutation spectrum) and their distribution across the genome, i.e., which genes and/or gene regulatory regions are affected. This may lead us to driver mutations, very similar to what has been discovered in cancers. In this respect we will be aided by the current state of the germ line heritable component of these disease, which may help us in identifying driver mutations. In turn, this may give us clues as to the development of interventions.
Deleterious effects due to random genomic changes that are not propagated through selection will be analyzed in a different way. As we previously showed (Bahar et al., 2006) and has now been amply confirmed by others (Enge et al., 2017), mRNA levels show increased cell-to-cell heterogeneity during aging. This could be caused by random somatic mutations in genes or gene-regulatory regions and maycause dysfunction and disease by interrupting protein-protein interactions. When the interaction of a protein with its partners is perturbed through a mutation, the protein is subjected to enhanced degradation as compared to that of its regular binding partners. We will test for that using multiplexed quantitative mass-spectrometry-based proteomics technology (Lapek et al., 2017).
However, it is also possible that increased mutation burdens act by accelerating certain age-related degenerative processes, most notably cellular senescence. Indeed, increased genome instability is a cause of senescence (Campisi and d'Adda di Fagagna, 2007) and there is ample evidence for a causal role of senescent cells in both atherosclerotic plaques (Matthews et al., 2006) and osteoarthritic lesions (Jeon et al., 2018).
All three potential mechanisms could be active in parallel. They could also lead to loss of connectivity among cells. For example, in atherosclerosis, endothelial cells must form tight connections and function in concert. One mutant cell could provide a break in essential connections causing a capillary or segment of an artery or vein to fail. For this reason we will also do connectivity analysis.
Read more
1. Bahar, R., Hartmann, C.H., Rodriguez, K.A., Denny, A.D., Busuttil, R.A., Dollé, M.E.T., Calder, R.B., Chisholm, G.B., Pollock, B.H., Klein, C.A., et al.(2006). Increased cell-to-cell variation in gene expression in aging mouse heart. Nature441, 1011-1014.
2. Beck, J.A., Poulter, M., Campbell, T.A., Uphill, J.B., Adamson, G., Geddes, J.F., Revesz, T., Davis, M.B., Wood, N.W., Collinge, J., et al.(2004). Somatic and germline mosaicism in sporadic early-onset Alzheimer's disease. Hum Mol Genet13, 1219-1224.
3. Bonanno, E., Mauriello, A., Partenzi, A., Anemona, L., and Spagnoli, L.G. (2000). Flow cytometry analysis of atherosclerotic plaque cells from human carotids: a validation study. Cytometry39, 158-165.
4. Campisi, J., and d'Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol8, 729-740.
5. DePinho, R.A. (2000). The age of cancer. Nature408, 248-254.
6. Dong, X., Zhang, L., Milholland, B., Lee, M., Maslov, A.Y., Wang, T., and Vijg, J. (2017). Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods14, 491-493.
7. Enge, M., Arda, H.E., Mignardi, M., Beausang, J., Bottino, R., Kim, S.K., and Quake, S.R. (2017). Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell171, 321-330 e314.
8. Erickson, R. (2003). Somatic gene mutation and human disease other than cancer. Mutation Research/Reviews in Mutation Research543, 125-136.
9. Fuster, J.J., MacLauchlan, S., Zuriaga, M.A., Polackal, M.N., Ostriker, A.C., Chakraborty, R., Wu, C.L., Sano, S., Muralidharan, S., Rius, C., et al.(2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science355, 842-847.
10. Jeon, O.H., David, N., Campisi, J., and Elisseeff, J.H. (2018). Senescent cells and osteoarthritis: a painful connection. J Clin Invest128, 1229-1237.
11. Lapek, J.D., Jr., Greninger, P., Morris, R., Amzallag, A., Pruteanu-Malinici, I., Benes, C.H., and Haas, W. (2017). Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat Biotechnol35, 983-989.
12. Lebedeva, A., Vorobyeva, D., Vagida, M., Ivanova, O., Felker, E., Fitzgerald, W., Danilova, N., Gontarenko, V., Shpektor, A., Vasilieva, E., et al.(2017). Ex vivo culture of human atherosclerotic plaques: A model to study immune cells in atherogenesis. Atherosclerosis267, 90-98.
13. Lodato, M.A., Rodin, R.E., Bohrson, C.L., Coulter, M.E., Barton, A.R., Kwon, M., Sherman, M.A., Vitzthum, C.M., Luquette, L.J., Yandava, C.N., et al.(2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science359, 555-559.
14. Matthews, C., Gorenne, I., Scott, S., Figg, N., Kirkpatrick, P., Ritchie, A., Goddard, M., and Bennett, M. (2006). Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res99, 156-164.
15. McCaffrey, T.A., Du, B., Consigli, S., Szabo, P., Bray, P.J., Hartner, L., Weksler, B.B., Sanborn, T.A., Bergman, G., and Bush, H.L., Jr. (1997). Genomic instability in the type II TGF-beta1 receptor gene in atherosclerotic and restenotic vascular cells. J Clin Invest100, 2182-2188.
16. Mertens, F., Palsson, E., Lindstrand, A., Toksvig-Larsen, S., Knuutila, S., Larramendy, M.L., el-Rifai, W., Limon, J., Mitelman, F., and Mandahl, N. (1996). Evidence of somatic mutations in osteoarthritis. Hum Genet98, 651-656.
17. Poduri, A., Evrony, G.D., Cai, X., and Walsh, C.A. (2013). Somatic mutation, genomic variation, and neurological disease. Science341, 1237758.
18. Vijg, J. (2014). Somatic mutations, genome mosaicism, cancer and aging. Curr Opin Genet Dev26C, 141-149.
19. Weakley, S.M., Jiang, J., Kougias, P., Lin, P.H., Yao, Q., Brunicardi, F.C., Gibbs, R.A., and Chen, C. (2010). Role of somatic mutations in vascular disease formation. Expert Rev Mol Diagn10, 173-185.
20. Zhang, L., and Vijg, J. (2018). Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu Rev Genet52, 397-419.
